Plant-Made Vaccines and Therapeutics
I have published a number of reviews on plant-made vaccines (see below), and our Biopharming Research Unit (affectionately known as “The BRU”) has been very active in this research area for nearly twenty years now. The theme running through all our publications is always “Plants are a cheaper, faster, safer and more scalable means of producing pharmaceutically-relevant proteins than any of the conventional expression systems…” Since 2003 we have published 50-odd articles on plant-made recombinant proteins, including human and animal vaccine proteins and enzymes, so we have used this justification a lot.
Which begs the question, why isn’t Big Pharma using plant plant production, then?
After all, it’s been 30-odd years since the first “molecular farming” product was made, and many proofs of principle and several of efficacy of therapeutics and vaccines have been obtained, yet the pharmaceutical world has just two products that have been licenced or emergency licenced for use in humans. The first is Elelyso from Pfizer, better known in molecular farming circles as glucocerebrosidase developed by Protalix, which is an enzyme replacement therapeutic for persons suffering from Gaucher disease. This is not strictly speaking a plant product, though, as it is made in transgenic suspension cultured carrot cells, in 800 litre plastic bags.
The other is ZMapp, which is a cocktail of three “humanised” monoclonal antibodies (mAbs) which bind to Ebola virus, made by transient expression in Nicotiana benthamiana plants, and which were used in people as a post-infection therapy in the West African Ebola disease outbreak from 2014-2016.
If you consider that the first products to receive regulatory body approval – both in 2006 – were a mAb to hepatitis B virus surface antigen (HBsAg) that was used in purification by a Cuban company of the protein from yeast culture lysates, and tobacco suspension culture-produced Newcastle disease virus vaccine made for Dow AgroSciences that was never marketed, there has been effectively no market breakout at all for plant-made pharmaceuticals (PMPs).
Why is this? Why is it that a technology that can produce biomass containing product-of-interest between 100 and 1000 times more cheaply than mammalian CHO cells, or 10 – 100-fold cheaper than yeast or bacterial cultures, and be scaled from lab to industrial levels of production quicker than any other system, still languishing in the biotech industry doldrums?

Rybicki, 2009: Drug Discov Today. 2009 Jan;14(1-2):16-24. doi: 10.1016/j.drudis.2008.10.002
Granted, biomass production is only the upstream part of pharmaceutical production; the downstream purification / refinement / vialling and packaging costs for plant-made products will be the same as for conventionally-made versions, and these are typically much higher than biomass production costs. My own back-of-the-envelope calculations, done at a conference I attended where these costs were broken down by an industry expert, came out with plant-made finished product in a vial being 32% cheaper than the conventional equivalent. Given the large markup on finished product, this “advantage” is in itself not sufficient motivation for Big Pharma to change the means of production, given their typically enormous investments in stainless steel and other infrastructure.
And yet…doubling production capacity for any given product by a single Big Pharma supplier using conventional cell culture technology would entail spending the same amount again to get more stainless steel – which is typically multiples of at least US$100 million – as well as spending an inordinately long time getting the new plant certified. Also, even making a new product from scratch using existing infrastructure would involve heroic cleaning and rejigging of tanks and feed pipes and other paraphernalia used for biomass production, recertifications and the like, which could take months. With plant-based manufacture, on the other hand, doubling production capacity means using double the number of cheaply-grown plants, possibly doubling the volume of Agrobacterium tumefaciens suspension to dunk them into, and then having enough space to put them under lights for 5-7 days or so, all with the same downstream processing capacity.
Then, there is the speed of scalability, which is unmatched for plant-made proteins. Consider this: given a ready supply of plants, it is theoretically possible for a molecular farming industrial facility to scale plant production of any given protein from lab bench scale – say a few milligrams/batch – to industrial scale (kilograms per batch), in as long a time it takes to culture the few hundred litres of Agrobacterium you would need for infiltration. Keeping a large reserve of plants is cheap; commercial greenhouses could do this very cheaply – meaning biomass is effectively instantly available to whatever volume required. Culturing Agrobacterium to scale would also literally take a couple of days, meaning infiltrating and incubation for target molecule synthesis could take just a few days from obtaining a gene. Scaling a new line of stably transfected CHO cells from a flask up to 30 000 litres, on the other hand…this takes many cell doublings, with the attendant problems of maintaining both genetic integrity and sterility, and is far more expensive and takes longer.
Plant-Made COVID-19 / SARS-CoV-2 Vaccines
In fact, in 2012 as part of the DARPA “Blue “Angel” challenge, Medicago Inc. of Quebec in their new North Carolina facility, managed to make 10 million doses of H1N1 influenza virus vaccine as virus-like particles (VLPs), vialled and labelled, within a month of being given the sequence of the virus. If one considers that seasonal influenza vaccines take at least six months to make by egg culture, even with accelerated clinical testing and certification, this is a truly impressive improvement on current technology, and probably the quickest development of an influenza virus vaccine ever*. The company has since advanced to making and testing a quadrivalent seasonal influenza vaccine candidate through Phase III clinical trial, for imminent commercial release, was awarded “Best New Vaccine Technology/Platform” prize at the World Vaccine Congress in 2019 – and on March 12th 2020 announced they had made a viable vaccine candidate against COVID-19. They did this in just 20 days after receiving (presumably) the S envelope glycoprotein gene, and moreover made VLPs using their proprietary technology: VLPs are better immunogens than soluble subunit proteins, as they are much better at stimulating both antibody and cellular immune responses.

Virus-like particles made the same way Medicago will probably make SARS-CoV-2 VLPs – from this paper: https://zoom.us/j/313676518?pwd=bnFrQmxtR3l2TjY4VGFWWEhjZklnZz09

 They are not alone in this space: just two weeks later, British American Tobacco (BAT) gained a lot of media attention when they also announced a candidate plant-made vaccine against SARS-CoV-2. While many hailed the repurposing of tobacco by a cigarette-manufacturing company as being an unexpected and good thing, it was really the BAT subsidiary RJ Reynolds’ recent purchase of Kentucky BioProcessing Inc, itself a spinout of one of the pioneering molecular farming companies (Large Scale Biology Inc., now sadly defunct) and the firm that had produced the largest amounts of the anti-Ebola ZMapp mAbs, that allowed them to take the credit. History aside, KBP announced they had “cloned a portion of COVID-19’s genetic sequence to create an antigen, which induce an immune response in the body” – which almost certainly means the S glycoprotein, or a portion of it – and that they could potentially make 3 million doses a week.
They are not alone in this space: just two weeks later, British American Tobacco (BAT) gained a lot of media attention when they also announced a candidate plant-made vaccine against SARS-CoV-2. While many hailed the repurposing of tobacco by a cigarette-manufacturing company as being an unexpected and good thing, it was really the BAT subsidiary RJ Reynolds’ recent purchase of Kentucky BioProcessing Inc, itself a spinout of one of the pioneering molecular farming companies (Large Scale Biology Inc., now sadly defunct) and the firm that had produced the largest amounts of the anti-Ebola ZMapp mAbs, that allowed them to take the credit. History aside, KBP announced they had “cloned a portion of COVID-19’s genetic sequence to create an antigen, which induce an immune response in the body” – which almost certainly means the S glycoprotein, or a portion of it – and that they could potentially make 3 million doses a week.
These announcements are the most important in the molecular farming space – although there have been others, such as by my long-time friend George Lomonossoff in the UK – and the vaccine candidates are almost certainly going to be cheaper and quicker to make than conventionally manufactured subunit-based equivalents like the Coalition for Epidemic Preparedness Innovations (CEPI)-sponsored University of Queensland product announced recently. Indeed, a local “futurist” – Pieter Geldenhuys, interviewed by Moneyweb on 29th March – said, of the news that Medicago had developed a vaccine:
“Once one of the multitude of medical research teams have developed an effective vaccine for the strain prevalent in South Africa, it will take several months, or even years, before enough vaccines could be produced to fill the global need. This is where tobacco plants come in”
Geldenhuys’s advice for various governments around the world is clear. Keep your ear on the ground and start reaching out to companies like these. Once initial tests show success, consider building your own tobacco cultivation plants to ensure that you can reproduce the vaccine at speed.
A very recent article in the Wall Street Journal also soberly assesses the prospects of plant-made vaccines against SARS2 – with some help from some molecular farmers we may know B-)
Molecular Farming Manufacturing Possibilities in South Africa
Our group in the BRU, our recent spinout partners* Cape Bio Pharms, and a group at the SA Council for Scientific and Industrial Research (CSIR) are the three premier molecular farming research and development teams in South Africa. We have jointly made a host of candidate vaccines, virus-derived reagents for use in molecular biology labs and in diagnostics, and mAbs for use as reagents and potentially as therapeutics. Presently, Cape Bio Pharms and possibly the CSIR represent the only pilot-scale manufacturing facilities in South Africa for plant-made biologics, despite initiatives over years involving us and the CSIR and various government departments. A symposium in Franschhoek in the Western Cape Province in November 2017, hosted by the BRU and by iBio Inc of Bryan Texas, pitched a plan to assembled invited delegates for public/private partnership to construct a facility in this country to make pharmaceutical products using molecular farming technology. In announcing it, we said the following:
iBio’s plant growth facility, October 2018
“The conference brings together leaders from public agencies, academic institutions, parastatals, private companies, regulators and private capital to map out concrete steps to establish the plant-based manufacturing platform in South Africa. The Department of Science and Technology (DST) leads a broad science and technology innovation effort including of advanced health care products to create socio-economic opportunities. The Technology Innovation Agency (TIA) is an active funder of human and animal health care initiatives in South Africa. The Industrial Development Corporation (IDC) is a primary developer of manufacturing capacity and has important initiatives in biotechnology. Other participating agencies include the Council for Scientific and Industrial Research (CSIR), with its own molecular farming pipeline, and the Department of Trade and Industry (DTI).
AzarGen Biotechnologies is a private South African biotechnology company will be part of the private sector representation. AzarGen, primarily funded by the IDC, has worked with iBio for the last three years to develop biotherapeutics that include surfactin for infant respiratory distress syndrome and a biobetter rituximab monoclonal antibody for the treatment of non-Hodgkin’s lymphoma and certain autoimmune diseases. The BioVac Institute and Onderstepoort Biologicals, manufacturers of human and animal vaccine products respectively, will also present. ENSafrica will speak to Intellectual Asset Management and Cape Venture Partners will overview the private capital opportunities in South Africa. Technology Innovation Group, a US based consulting group, will talk about the structure of successful public/private partnerships.”

While the idea of a full-scale facility similar to iBio’s – costed at around USD30 million/R450 million – did not appeal to funders present, the idea of a cGMP-certified pilot manufacturing facility costing USD10 million – R150 million at the time – constructed using iBio’s expertise and assistance, found more favour. In fact, various entities promised to survey interested parties to establish the need and feasibility of internally funding it.
To the best of my knowledge, nothing along the lines of a survey has happened to date. Since then, and in the absence of any apparent interest from what were DST, DTI, IDC, TIA and others, iBio has gone on in 2019 to announce a partnership with Azargen in the area of rituximab biosimilar production, and as of a few days ago, as a contract manufacturing organisation, is offering their services in making COVID/SARS2 reagents at industrial scale in plants. Cape Bio Pharms has also established itself as a reagent manufacturer independently of any outside associations, with only local investment and a THRIP grant from Dept of Trade and Industry (DTI). I note that a previous proposal from some years ago involving the CSIR and Kentucky BioProcessing for establishment of an even cheaper pilot facility, also fell flat. For comparison, I will point out that the cost of just the revamping of Onderstepoort Biological Products’ (OBP, SA’s premier veterinary vaccine manufacturer) facility to be able to achieve cGMP certification is estimated to be ~R500 million.
SARS2/COVID Vaccines and Reagents for South Africa
Very early on in the present pandemic, Dr Mani Margolin of both the BRU and the Vaccine Research Group (VRG) of Prof Anna-Lise Williamson ordered a synthetic gene for a soluble version of the SARS-CoV-2 S protein, and has since successfully expressed the protein in both tissue cultured human cells, and in Nicotiana benthamiana plants via transient Agrobacterium-mediated expression. Both expression strategies leveraged technologies for which our research groups have either applied for or been granted patents, and established the very real possibilities of making both a DNA vaccine and a protein subunit vaccine against SARS2. He has gone on to insert the S protein gene into other vaccine vectors in the VRG.
Cape Bio Pharms (CBP)*, acting in parallel, ordered a gene for the “head” portion of the S protein – termed S1 – which they have also successfully expressed in N benthamiana, along with several variants of the protein, and they plan to collaborate with another new biotech company in South Africa to use it to produce mAbs for use as reagents, and potentially as therapeutics.
The CSIR is planning to leverage their established expertise in making mAbs to HIV and rabies in plants to produce a panel of mAbs to SARS2 for the same purposes.
These efforts have already resulted in ad hoc partnerships with other research groups and organisations, with S and S1 protein being supplied to others for use in establishing enzyme immunoassays and other diagnostic tests for serosurveillance and bedside testing, and other genes being shared with us and CBP for expression as reagents. I will note that the efforts that have resulted in the S-derived products are probably the fastest production at scales greater than a few micrograms in this country of any protein-based reagents, and probably the most quickly and cheaply scalable of any reagents. We are presently awaiting news of possible funding for molecular farming projects involving SARS2, albeit in a very rapidIy changing landscape where every day brings new developments – and where the future economic prospects of our country look dire, which may work against us.
Lessons From the Past
We have been here before, though. In 2006 our group received “Emergency Response” 1-year funding for H5N1 vaccine development from the Poliomyelitis Research Foundation (PRF) in SA – a then-handsome amount of R250 000 – which we then parlayed into another PRF 3-year grant, as well funding from the SA Medical Research Council (SAMRC). This quote from a profile published in Human Vaccines & Immunotherapeutics nicely sums up what we did:
As a result of a conference held in Cape Town in 2005, where a WHO influenza expert warned us “When the pandemic comes, you in the developing countries will be on your own”, we applied for extraordinary funding from the PRF in SA to explore the possibility of making a pandemic flu virus vaccine in South Africa. We chose the highly pathogenic avian influenza virus A H5N1 type haemagglutinin (H5 HA) as a target, and James Maclean was again instrumental in designing and successful early testing of plant-made soluble and membrane-bound forms. Further funding from the PRF and the SA MRC allowed proof of principle that we could in fact produce flu virus vaccine candidates in South Africa – both as [plant-made] subunit protein and as DNA vaccines.
In retrospect, while these projects were impossibly ambitious and not a little naïve, we and our co-workers received a crash course in both research vaccinology and the handling of big projects that has been crucial for all our subsequent work. We were also able to establish stable and well-qualified teams of people, with a nucleus of senior scientists who have been around us for up to 15 years. Another very important lesson was that we should patent our discoveries: in my case, this has led to me and my co-workers having the largest patent portfolio at our institution, and the largest molecular biotechnology-related portfolio in Africa – most of them to do with vaccines (14+ patent families). The development of a set of well-tried protocols around expression of novel antigens in a variety of systems has also been invaluable – especially when funding circumstances demanded that we change direction….
The potential importance of molecular farming for human health has been underlined recently with the apparently successful use of plant-produced MAbs (ZMapp) against Ebola virus disease in West Africa, and the proof of large-scale and rapid emergency-response production in plants of potentially pandemic influenza vaccines by Medicago Inc, among others [my emphasis] . We see our future role in exploiting niche opportunities for production of vaccine candidates and reagents for orphan or geographically-limited disease agents that do not attract Big Pharma attention – like CCHFV and RVFV – as well as for emerging animal diseases such as BTV and AHSV and BFDV, where rapid responses and small manufacturing runs may be needed [my emphasis].
Despite the fact that we ambitiously entitled our 2012 flu vaccine paper “Setting up a platform for plant-based influenza virus vaccine production in South Africa“, and our 2013 DNA vaccine paper as “An H5N1 influenza DNA vaccine for South Africa“, nothing happened. Nothing, despite the then Minister of Health Dr Aaron Motsoaledi saying during the influenza H1N1 2009 pandemic, that:
“South Africa has arrived at a situation where we have no option but to start developing our own vaccine capacity, not only for H1N1, but generally,” Motsoaledi told parliament.
“The disturbing feature about today’s world… has been expressed by the minister of health for Cambodia… who noted that the developed world, after producing the vaccine, may want to cover their own population first before thinking about the developing world,” Motsoaledi said.
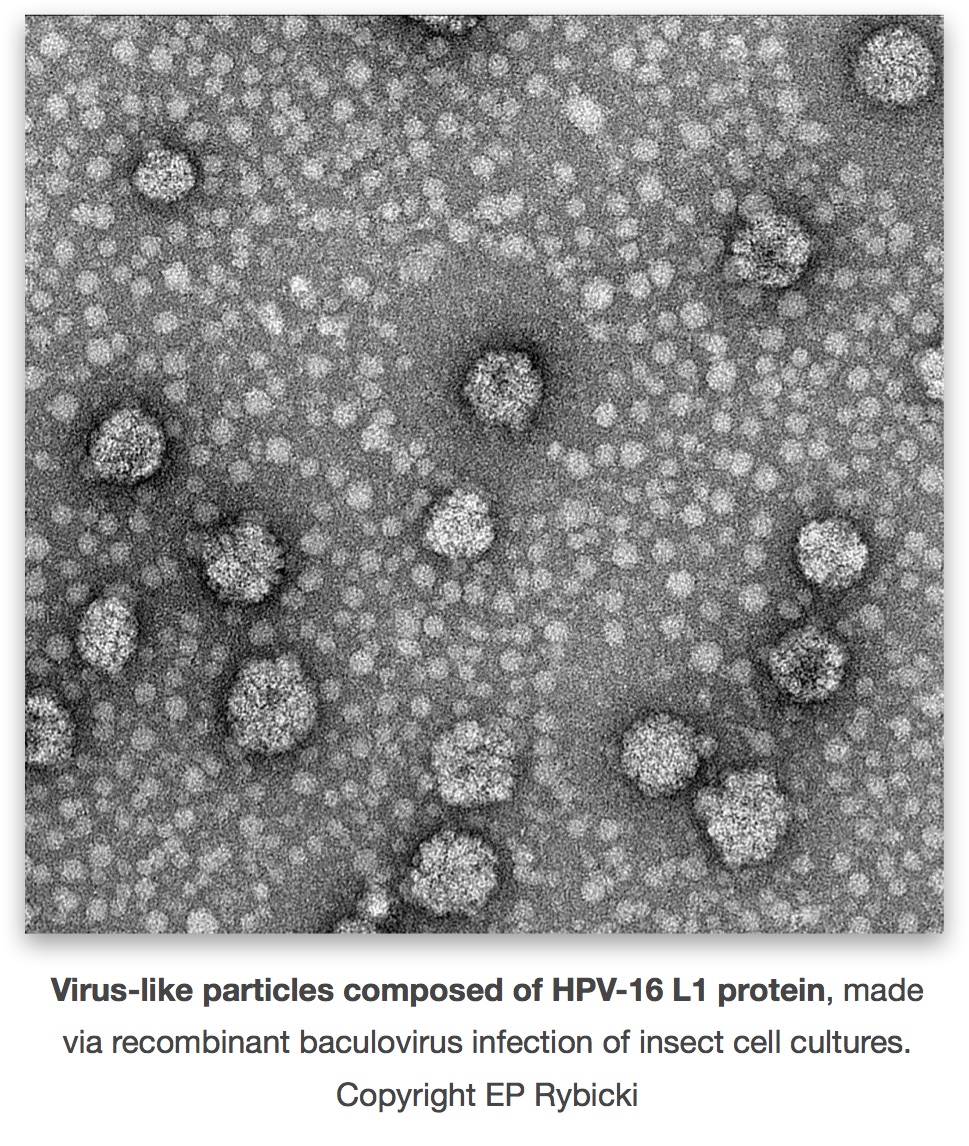
It’s been nearly 11 years. Nothing has happened still. Despite distributing some 25 million doses of vaccines annually in South Africa, our only human vaccine firm – The Biovac Institute – still makes no virus vaccines. We have licenced our patented technology – for plant-made human papillomavirus vaccines and influenza virus vaccine – outside the country, for the lack of any interest locally.
This really should change. Maybe we have an opportunity now.
*= potential conflicts of interest due to partnerships.
Reviews on Molecular Farming
1: Dennis SJ, Meyers AE, Hitzeroth II, Rybicki EP. African Horse Sickness: A Review of Current Understanding and Vaccine Development. Viruses. 2019 Sep 11;11(9). pii: E844. doi: 10.3390/v11090844. Review. PubMed PMID: 31514299; PubMed Central PMCID: PMC6783979.
2: Rybicki EP. Plant molecular farming of virus-like nanoparticles as vaccines and reagents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2020 Mar;12(2):e1587. doi: 10.1002/wnan.1587. Epub 2019 Sep 5. Review. PubMed PMID: 31486296.
3: Chapman R, Rybicki EP. Use of a Novel Enhanced DNA Vaccine Vector for Preclinical Virus Vaccine Investigation. Vaccines (Basel). 2019 Jun 13;7(2). pii: E50. doi: 10.3390/vaccines7020050. Review. PubMed PMID: 31200559; PubMed Central PMCID: PMC6632145.
4: Margolin E, Chapman R, Williamson AL, Rybicki EP, Meyers AE. Production of complex viral glycoproteins in plants as vaccine immunogens. Plant Biotechnol J. 2018 Jun 11. doi: 10.1111/pbi.12963. [Epub ahead of print] Review. PubMed PMID: 29890031; PubMed Central PMCID: PMC6097131.
5: Chabeda A, Yanez RJR, Lamprecht R, Meyers AE, Rybicki EP, Hitzeroth II. Therapeutic vaccines for high-risk HPV-associated diseases. Papillomavirus Res. 2018 Jun;5:46-58. doi: 10.1016/j.pvr.2017.12.006. Epub 2017 Dec 19. Review. PubMed PMID: 29277575; PubMed Central PMCID: PMC5887015.
6: Rybicki EP. Plant-made vaccines and reagents for the One Health initiative. Hum Vaccin Immunother. 2017 Dec 2;13(12):2912-2917. doi: 10.1080/21645515.2017.1356497. Epub 2017 Aug 28. Review. PubMed PMID: 28846485; PubMed Central PMCID: PMC5718809.
7: Williamson AL, Rybicki EP. Justification for the inclusion of Gag in HIV vaccine candidates. Expert Rev Vaccines. 2016 May;15(5):585-98. doi: 10.1586/14760584.2016.1129904. Epub 2015 Dec 28. Review. PubMed PMID: 26645951.
8: Rybicki EP. Plant-based vaccines against viruses. Virol J. 2014 Dec 3;11:205. doi: 10.1186/s12985-014-0205-0. Review. PubMed PMID: 25465382; PubMed Central PMCID: PMC4264547.
10: Scotti N, Rybicki EP. Virus-like particles produced in plants as potential vaccines. Expert Rev Vaccines. 2013 Feb;12(2):211-24. doi: 10.1586/erv.12.147. Review. PubMed PMID: 23414411.
11: Thuenemann EC, Lenzi P, Love AJ, Taliansky M, Bécares M, Zuñiga S, Enjuanes L, Zahmanova GG, Minkov IN, Matić S, Noris E, Meyers A, Hattingh A, Rybicki EP, Kiselev OI, Ravin NV, Eldarov MA, Skryabin KG, Lomonossoff GP. The use of transient expression systems for the rapid production of virus-like particles in plants. Curr Pharm Des. 2013;19(31):5564-73. Review. PubMed PMID: 23394559.
12: Rybicki EP, Hitzeroth II, Meyers A, Dus Santos MJ, Wigdorovitz A. Developing country applications of molecular farming: case studies in South Africa and Argentina. Curr Pharm Des. 2013;19(31):5612-21. Review. PubMed PMID: 23394557.
14: Lotter-Stark HC, Rybicki EP, Chikwamba RK. Plant made anti-HIV microbicides–a field of opportunity. Biotechnol Adv. 2012 Nov-Dec;30(6):1614-26. doi: 10.1016/j.biotechadv.2012.06.002. Epub 2012 Jun 28. Review. PubMed PMID: 22750509.
15: Rybicki EP, Martin DP. Virus-derived ssDNA vectors for the expression of foreign proteins in plants. Curr Top Microbiol Immunol. 2014;375:19-45. doi: 10.1007/82_2011_185. Review. PubMed PMID: 22038412.
16: Rybicki EP, Chikwamba R, Koch M, Rhodes JI, Groenewald JH. Plant-made therapeutics: an emerging platform in South Africa. Biotechnol Adv. 2012 Mar-Apr;30(2):449-59. doi: 10.1016/j.biotechadv.2011.07.014. Epub 2011 Aug 3. Review. PubMed PMID: 21839824.
17: Rybicki EP, Williamson AL, Meyers A, Hitzeroth II. Vaccine farming in Cape Town. Hum Vaccin. 2011 Mar;7(3):339-48. Epub 2011 Mar 1. Review. PubMed PMID: 21358269.
18: Giorgi C, Franconi R, Rybicki EP. Human papillomavirus vaccines in plants. Expert Rev Vaccines. 2010 Aug;9(8):913-24. doi: 10.1586/erv.10.84. Review. PubMed PMID: 20673013.
19: Rybicki EP. Plant-made vaccines for humans and animals. Plant Biotechnol J. 2010 Jun;8(5):620-37. doi: 10.1111/j.1467-7652.2010.00507.x. Epub 2010 Mar 11. Review. PubMed PMID: 20233333.
20: Pereira R, Hitzeroth II, Rybicki EP. Insights into the role and function of L2, the minor capsid protein of papillomaviruses. Arch Virol. 2009;154(2):187-97. doi: 10.1007/s00705-009-0310-3. Epub 2009 Jan 25. Review. PubMed PMID: 19169853.