This is an update of a post I did on Alan Cann’s MicrobiologyBytes back in 2007, before i started ViroBlogy: I am doing this because (a) it’s mine, (b) I want to update it – and the MB version is archived, so I can’t. So here we are again:
I think it’s permissible, after working on your favourite virus for over 20 years, to develop some sort of feeling for it: you know, the kind of insight that isn’t directly backed up by experiment, but that may very well be right. Or not – but in either case, it would take a deal of time and a fair bit of cash to prove or disprove, and would have sparked some useful discussion in the meantime. And then, of course, the insights you have into (insert favourite virus name here) – if correct – can usually be extended into the more general case, and if you are sufficiently distinguished, people may actually take them on board, and you will have contributed to Accepted Wisdom.
I can’t pretend – at least, outside of my office – to any such Barbara McClintock-like distinction; however, I have done a fair bit of musing on my little sphere of interest as it relates (or not) to the State of the Viral Universe, and I will share some of these rambles now with whomever is interested.
I have been in the same office now, and teaching the same course, more or less, for 32-odd years. In that time I have worked on the serology and epidemiology of the bromoviruses, cucumovirus detection, potyvirus phylogeny, geminivirus diversity and molecular biology, HIV and papillomavirus genetic diversity, and expressing various bits of viruses and other proteins in plants and in insect cells. However, much of my interest (if not my effort) in that time has been directed towards understanding how grass-infecting mastreviruses in particular interact with their environment and with each other, in the course of their natural transmission cycle.

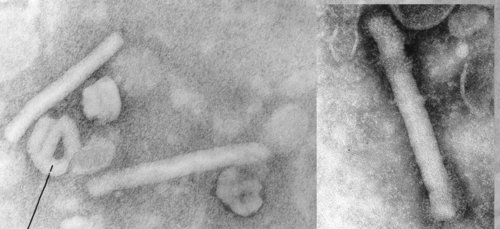
Maxwell’s Demon (left, lower) and Martian Face (right, upper) visible on a MSV virion
Fascinating little things, mastreviruses: unique geminate capsid architecture, and at around a maximum of 2.8 kb of single-strand circular DNA, we thought they were the smallest DNA genomes known until the circoviruses and then the zoo of anello- and anello-like viruses were discovered. Their genomes code for only 4 proteins – two replication-associated, one movement and one capsid – yet we have managed to work on just one subgroup of mastrevirus species for 27 years, without exhausting its interest – at least, to us… (see PubMed list here). We also showed that one could see Martian faces quite distinctly on virions – and possibly even Maxwell’s Demon. But I digress….

Severe symptoms of MSV on sweetcorn
We have concentrated on the “African streak viruses” – related species Maize streak virus, Panicum streak virus, Digitaria streak virus, Sugarcane streak virus and friends – for two very simple reasons:
1. They occur in Africa, near us, and nowhere else;
2. Maize streak virus is the worst viral pathogen affecting maize in Africa.
So we get situational or niche advantage, and we get to work on an economically-important pathogen. One that was described – albeit as “…not of…contagious nature” – as early as 1901, no less.

Maize streak virus or MSV, like its relatives, is obligately transmitted by a leafhopper (generally Cicadulina mbila Naudé): this means we have a three-party interaction – of virus-host-vector – to consider when trying to understand the dynamics of its transmission. Actually, it’s more complicated than that: we have also increasingly to consider the human angle, given that the virus disease affects mainly the subsistence farming community in Africa, and that human activity has a large influence on the spread of the disease. So while considering just the virus – as complicated as that is – we have to remember that it is only part of the whole picture.
So how complicated is the virus? At first sight, not very: all isolates made from severe maize infections share around 97% of their genome sequence. However, however…that 3% of sequence variation hides a multitude of biological differences, and there is a range of relatives infecting grasses of all kinds, some of which differ by up to 35% in genome sequence. Moreover, maize is a crop plant first introduced to Africa a maximum of 500 years ago, so it is hardly a “natural” host – yet, all over Africa, it is infected by only a very narrow range of virus genotypes, from a background of very wide sequence diversity available.
So here’s an insight:
the host selects the virus that replicates best in it.
And lo, we found that in the Vaalharts irrigation area in the north of South Africa that the dominant virus genotype in winter wheat was a different strain – >10% sequence difference – to the one in the same field, in summer maize. Different grass species also have quite different strains or even species of streak viruses best adapted to them.
 Not all that profound a set of observations, perhaps, but they lead on to another insight:
Not all that profound a set of observations, perhaps, but they lead on to another insight:
streak viruses travel around as a cloud of variants or virus complex.
Not intuitively obvious, perhaps…but testable, and when we did, we found we were right: cloning virus genomes back out of maize or from a grass infected via leafhoppers gave a single predominant genotype in each case, with a number of other variants present as well. Looking further, we discovered that even quite different viruses could in fact trans-replicate each other: that is, the Rep/RepA complex of one virus could facilitate the replication of the genome of a virus differing by up to 35% in DNA sequence. We have also – we think – made nonsense of the old fancy that you could observe “host adaptation” of field isolates of MSV: we believe this was due to repeated selection by a single host genotype from the “cloud” of viruses transmitted during the natural infection cycle.
So, insight number three:
there is a survival benefit for the viruses in this strategy.
This is simple to understand, really, and relates to leafhopper biology as well as to host: the insects move around a lot, chasing juicy grasses, and it would be an obvious advantage to the streak virus complex to be able to replicate as a complex in each different host type – given that different virus genotypes have differential replication potential in the various backgrounds. This is quite significantly different, incidentally, to what happens with the very distantly-related (in terms of geological time) begomoviruses, or whitefly-transmitted geminiviruses: these typically do not trans-replicate each other across a gap of more than 10% of sequence difference.
Boring, I hear you say, but wait…. Add another factoid in, and profound insights start to emerge. In recent years, the cloud of protégés or virologist complex around me has accumulated to critical mass, and one of the most important things to emerge – apart from some frighteningly effective software for assessing recombination in viral genomes, which I wish he’d charge for – was Darren Martin’s finding that genome recombination is rife among African streak viruses. This was unexpected, given the expectation that DNA viruses simply don’t do that sort of thing; that promiscuous reassortment of components between genomes is a hallmark of RNA viruses. Makes sense in retrospect (an exact science), however, because of the constraints on DNA genomes: how else to explore sequence space, if the proof-reading is too good? And if you travel in a complex anyway…why not swap bits for biological advantage?

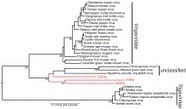
Linkage map of the MSV genome, showing what interacts with what
So Darren swapped a whole lot of bits, in a tour-de-force of molecular virology, to create some 54 infectious chimaeric MSV genomes – and determined that
“The pathogenicity of chimeras was strongly influenced by the relatedness of their parental viruses and evidence was found of nucleotide sequence-dependent interactions between both coding and intergenic regions“.
In other words –new insight:
the whole genome is a pathogenicity determinant, and bits of it interact with other bits in unexpected ways.
At this point you could say “Hey, all his insights are in fact hypotheses!” – and you would be partially correct, except for
Profound Insight No. 1: hypotheses are the refuge of the linear-thinking.
Or its variant, found on my office wall:
“**c* the hypotheses, let’s just discover something”. I also have
“If at first you don’t succeed, destroy all evidence that you tried” and a number of exotic beer bottle labels on my wall – but I digress….
As an aside here, I am quite serious in disliking hypothesis-driven science: I think it is a irredeemably reductionist approach, which does not easily allow for Big Picture overviews, and which closes out many promising avenues of investigation or even of thought. And I teach people how to formulate them so they can get grants and publications in later life, but I still think HDS is a tyranny that should be actively subverted wherever possible.
Be all this as it may, now follows
Profound Insight No. 2: genome components may still be individually mobile even when covalently linked.
Now take a moment to think on this: recombination allows genes to swap around inside genetic backgrounds so as to constitute novel entities – and the “evolutionary value of exchanging a genome fragment is constrained by the number of ways in which the fragment interacts with the rest of the genome*“. Whether or not the genome is RNA, DNA, in one piece or divided. All of a sudden, the concept of a “virus genome” as a gene pool rather than a unitary thing becomes obvious – and so does the reductionism inherent in saying “this single DNA/RNA sequence is a virus”.
So try this on for size for a brand-new working definition of a virus – and
Profound Insight No. 3: a virus is an infectious acellular entity composed of compatible genomic components derived from a pool of genetic elements.
Sufficiently paradigm-shifting for you? Compare it to more classical definitions – yes, including one by AJ Cann, Esq. – and see how much simpler it is. It also opens up the possibility that ANY virus as currently recognised is simply an operational assembly of components, and not necessarily the final article at all.
Again, my favourite organisms supply good object examples: the begomoviruses – whitefly-transmitted geminiviruses –
- may have one- or two-component genomes;
- some of the singleton A-type components may pick up a B-type in certain circumstances;
- some doubletons may lose their B without apparent effect in model hosts;
- some A components may apparently share B components in natural infections;
- the A and B components recombine like rabbits with cognate molecules (or Bs can pick up the intergenic region from As);
- in many cases have one or more satellite ssDNAs (β DNA, or nanovirus-related components) associated with disease causation;
…and so on, and on…. An important thing to note here is the lab-rat viruses – those isolated early on, and kept in model plant species in greenhouses – often don’t exhibit any of these strangenesses, whereas field-isolated viruses often do.
Which tells you quite a lot about model systems, doesn’t it?
But this is not only true of plant viruses: the zoo of ssDNA anello-like viruses found in humans and in animals – with several very distantly-related viruses to be found in any individual, and up to 80% of humans infected – just keeps on getting bigger and weirder. Added to the original TT virus – named originally for the initials of the Japanese patient from whom it was isolated, and in a post hoc exercise of convoluted logic, named Torque teno virus (TTV) [why don’t people who work with human or animal viruses obey ICTV rules??] – are now Torque teno minivirus (TTMV) and “small anellovirus” SAV) – all of which have generic status. And all of which may be the same thing – as in, TTVs at a genome size of 3.63.8 kb may give rise to TTMVs (2.8-29 kb) and SAVs (2.4-2.6 kb) as deletion mutants as part of a population cloud, where the smaller variants are trans-replicated by the larger. Thus, a whole lot of what are being described as viruses – without fulfilling Koch’s Postulates, I might point out – are probably only “hopeful monsters” existing only as part of a population. Funnily enough, this sort of thing is much better explored in the ssDNA plant virus community, given that working with plant hosts is so much easier than with human or animal.
And now we can go really wide, and attempt to be profound on a global scale: it should not have escaped your notice that the greatest degree of diversity among organisms on this planet is that of viruses, and viruses that are found in seawater in particular. There is a truly mind-boggling number of different viruses in just one ml of seawater taken from anywhere on Earth, which leads respectable authors such as Curtis Suttle to speculate that viruses almost certainly have a significant influence on not only populations of all other marine organisms, but even on the carbon balance of the world’s oceans – and therefore of the planet itself.
Which leads to the final, and most obvious,
Profound Insight (No. 4): in order to understand viruses, we should all be working on seawater….
That is where the diversity is, after all; that is where the gene pool that gave rise to all viruses came from originally – and who knows what else is being

Hypolith – cyanobacteria-derived, probably – under a piece of Namib quartzite from near Gobabeb Research Station
cooked up down there?
And this is the major update: not only have I managed to get funded for a project on “Marine Viromics” from our local National Research Foundation – a process akin to winning the lottery, and about as likely to succeed – I am also collaborating with friends and colleagues from the Institute for Microbial Biotechnology and Metagenomics at the University of the Western Cape on viruses in desert soils, and associated with hypoliths– or algal growths found under quartzite rocks in extreme environments.
Thus, I shall soon be frantically learning how to deal with colossal amounts of sequence data, and worse, learning how to make sense of it. We should have fun!
——————————————————————————————————————–
* And as a final curiosity, I find that while I – in common with the World Book Encyclop[a]edia and Learning Resources – take:mol|e|chism or mol|e|cism «MOL uh KIHZ uhm», noun. to mean any virus, viewed as an infective agent possessing the characteristics of both a living microorganism and a nonliving molecule; organule.
[molechism < mole(cule) + ch(emical) + (organ)ism; molecism < molec(ule) + (organ)ism] –
There is another meaning… something to do with sacrifice of children and burning in hellfire eternally. This is just to reassure you that this is not that.






{kind=link}